(Require version v1.3 or above)
The analysis may need following additional software:
- samtools >= v1.9.
- BWA >= v0.7.17.
- taiji-utils: install using
pip install taiji-utils.
Preparing input and configuration files
To start, create a TAB-delimited file named input.tsv and a YAML file named config.yml. Taiji accepts multiple types of input. Below are example input and configuration files for common input types. Detailed documentation about the input format can be found here.
File 1: input.tsv
type id rep path tags format
scATAC-seq pbmc_10k_nextgem 1 URL:https://cf.10xgenomics.com/samples/cell-atac/1.2.0/atac_pbmc_10k_nextgem/atac_pbmc_10k_nextgem_fragments.tsv.gz PairedEnd,Gzip Bed
scATAC-seq pbmc_10k_v1 1 URL:https://cf.10xgenomics.com/samples/cell-atac/1.1.0/atac_pbmc_10k_v1/atac_pbmc_10k_v1_fragments.tsv.gz PairedEnd,Gzip BedIn this example input file, we tell Taiji to download the fragment file from the 10x Genomics website.
File 2: config.yml
input: "input.tsv"
output_dir: "output/"
genome: "path-to-genome-fasta/GRCh37.fa"
annotation: "path-to-annotation/gencode.v34lift37.basic.annotation.gtf"
scatac_options:
cluster_optimizer: CPMSee here for all available options and their roles.
File 1: input.tsv
type id group rep path tags
scATAC-seq forebrain_E11.5 forebrain_E11.5 1 ENCFF951ZRW,ENCFF197HRD ENCODE,Demultiplexed
scATAC-seq forebrain_P0 forebrain_P0 1 ENCFF501RJM,ENCFF595BQU ENCODE,DemultiplexedIn this example we used demultiplexed FASTQ files from ENCODE, in which the FASTQ files were demultiplexed by adding the barcode to the beginning of each read in the following format: “@” + “barcode” + “:” + “read_name”. Below is one example of demultiplexed fastq file:
$ zcat CEMBA180306_2B.demultiplexed.R1.fastq.gz | head
@AGACGGAGACGAATCTAGGCTGGTTGCCTTAC:7001113:920:HJ55CBCX2:1:1108:1121:1892 1:N:0:0
ATCCTGGCATGAAAGGATTTTTTTTTTAGAAAATGAAATATATTTTAAAG
+
DDDDDIIIIHIIGHHHIIIHIIIIIIHHIIIIIIIIIIIIIIIIIIIIIIRemove the “ENCODE” tag if your files are in a local path, for example:
type id group rep path tags
scATAC-seq forebrain_E11.5 forebrain_E11.5 1 e11.5_R1.fastq.gz,e11.5_R2.fastq.gz Demultiplexed
scATAC-seq forebrain_P0 forebrain_P0 1 p0_R1.fastq.gz,p0_R2.fastq.gz DemultiplexedFile 2: config.yml
input: "input.tsv"
output_dir: "output/"
bwa_index: "path-to-BWAIndex/genome.fa"
genome: "path-to-genome-fasta/genome.fa"If you don’t have genome fasta file or BWA index on your computer, you can tell Taiji to automatically download that for you by specifying the genome assembly:
input: "input.tsv"
output_dir: "output/"
assembly: "mm10"Launch the preprocessing pipeline
(Omit this step if starting from the fragment files)
The preprocessing pipeline consists of reads mapping, reads filtering, quality control. Use the command below to execute the preprocessing pipeline:
taiji run --config config.yml --select SCATAC_Remove_DuplicatesA fragment file will be generated for each FASTQ input in the output directory. For the format of the fragment file, see here: https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/output/fragments.
Quality control
taiji run --config config.yml --select SCATAC_QCIn the SCATACSeq/QC directory, you will find QC results. Here are one example:
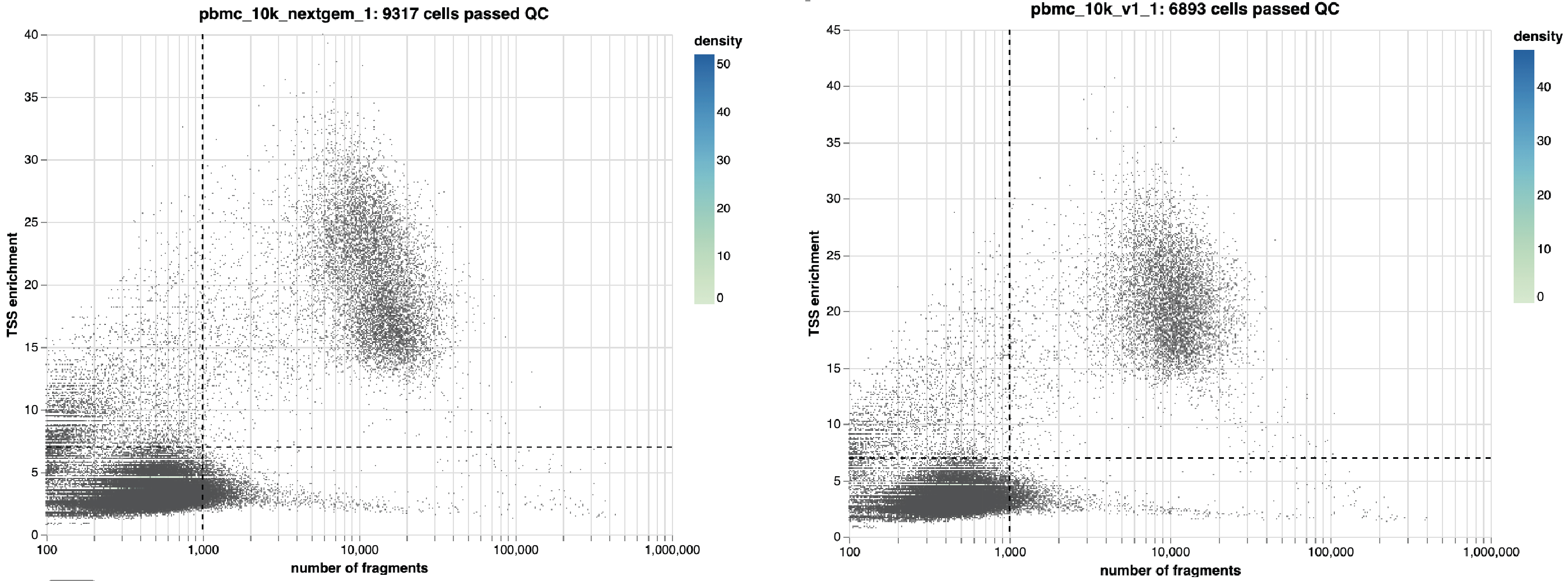
This figure shows the cells are filtered by the number of unique fragments (default is 1,000) and the TSS enrichment (default is 7). The cutoffs can be changed in the config.yml, for example:
scatac_options:
tss_enrichment_cutoff: 7
fragment_cutoff: 1000Joint clustering analysis
taiji run --config config.yml --select SCATAC_Merged_ClusterTo determine the number of clusters, Taiji will search over specified parameter values and optimize for cluster separation and cluster stability.
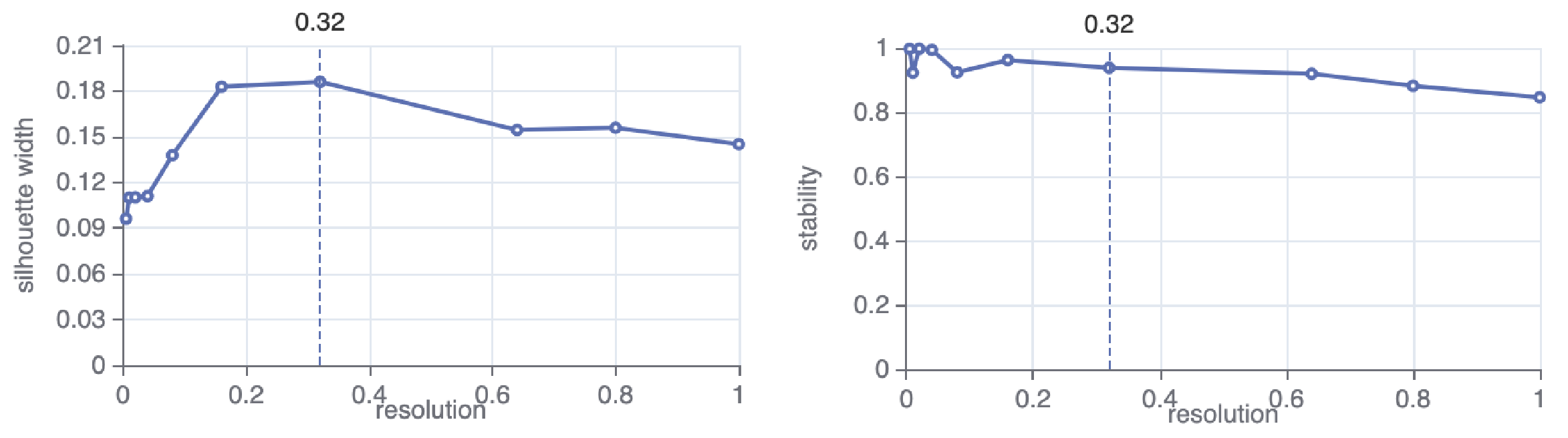
Based on the search result, Taiji will automatically select a parameter value (here is 0.32). You can change this as well as the parameter values to search for in the config file, for example:
scatac_options:
cluster_resolution_list: [0.001, 0.002, 0.02, 0.05, 0.1, 0.25, 1]
cluster_resolution: 0.5In SCATACSeq/Cluster/Merged_rep1_cluster.html, you can find a visualization of the clustering result.

More analyses
Use taiji view taiji.html to see what are availiable!