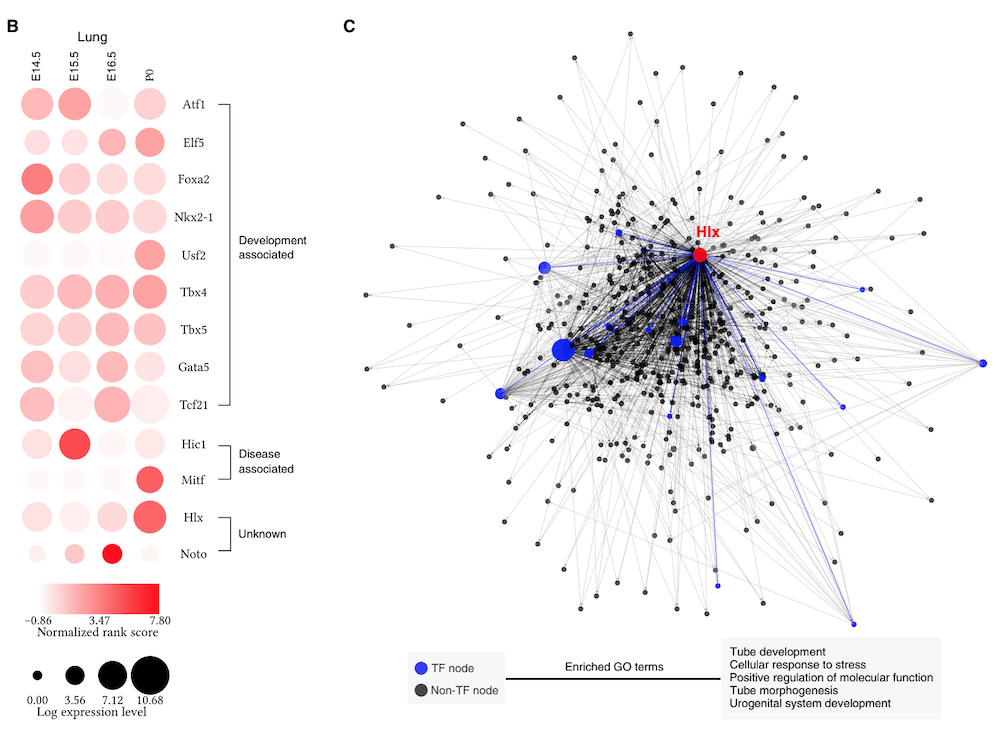
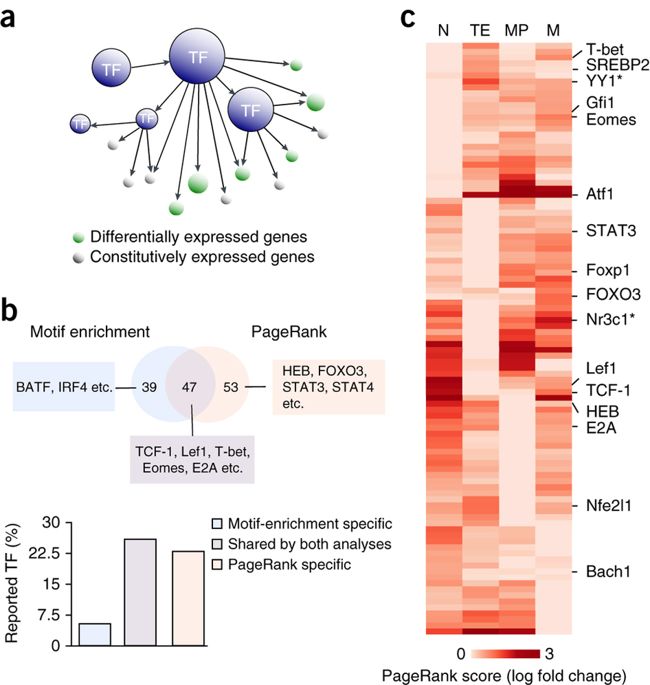
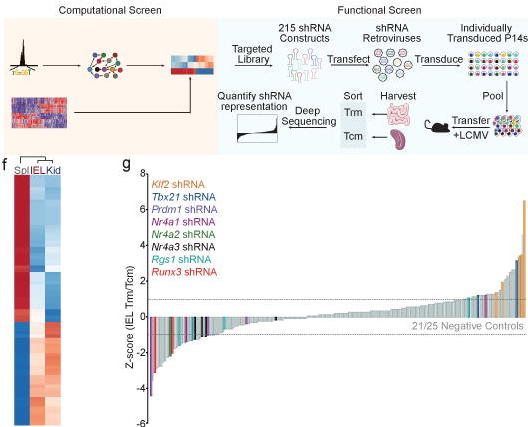
The Taiji software is an integrative multi-omics data analysis framework. It can be used as a standalone pipeline to analyze ATAC-seq, RNA-seq, single cell ATAC-seq or Drop-seq data. However, the uniqueness and the power of Taiji really lie in its ability to integrate diverse datasets and use these information in a clever way to construct regulatory network and identify candidate driver genes.
The design philosophy of the Taiji pipeline is focused on:
- Correctness: We only include reliable algorithms and make every effort to ensure the implementations are bug-free.
- Performance: We code algorithms from scratch when necessary to ensure the pipeline can scale to large datasets (thousands of samples at least).
- Convinence: Most analyses have multipe entry points, e.g., Fastq, Bam or Bed. The execution of the pipeline requires only a single command.
We achieve these at the expense of customization. This will be improved in the future.
Quick Start
input: "input.tsv"
output_dir: "output/"
assembly: "GRCh38"type id group rep path tags
ATAC-seq control control 1 ENCFF893KQZ,ENCFF443HVZ ENCODE
ATAC-seq 2h 2h 1 ENCFF173INV,ENCFF322IZC ENCODE
ATAC-seq 4h 4h 1 ENCFF562JHD,ENCFF943SYH ENCODEtaiji run --config config.yml -n 3 +RTS -N3
Tutorials
If you have used Taiji in your research, please consider cite the following paper:
K. Zhang, M. Wang, Y. Zhao, W. Wang, Taiji: System-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci. Adv. 5, eaav3262 (2019).